Cell therapy commercialization: GMP and Scalability

In this editorial, David Pettitt and David Brindley discuss the complex process of biomanufacturing and its importance in commercializing a cell therapy.
David Pettitt1 & David Brindley2
1Academic Surgeon & SENS Research Foundation Doctoral Scholar, University of Oxford, UK; [email protected]
2University of Oxford, UK; [email protected]
Recent advances in bioengineering have positioned cellular therapeutics at the cusp of what may be a transformational shift in the management of unmet medical needs, and have generated considerable excitement in clinical and scientific communities. Cell and gene-based therapies are exciting platform technologies with the capacity to facilitate the de novo production of functional tissue (Culme-Seymour, Davie et al. 2012, Zacharias, Nelson et al. 2011). Although a number of cell therapy products have achieved success across European Union (EU), United States (US) and global marketplaces, commercializing cellular based therapies remains challenging (Pettitt, Arshad et al. 2017). In part, this is secondary to the requisite for complex manufacturing procedures that are meticulously aligned with highly specific regulatory and quality requirements.
The process for generating cell therapies is complex and they require substantially different manufacturing approaches to conventional therapeutics (Brindley, Wall et al. 2013). A manufacturer’s approach is largely dictated by whether the intended therapeutic is allogeneic (from a single source or master cell bank) or autologous (isolated from a patient) (Digiusto DL, Kiem HP 2012). Allogeneic products are more amenable to scale-up manufacturing or an “off-the-shelf” business model whereas autologous-based products are more suited to scaled-out approaches due to their patient specificity and immuno-compatibility requirements (Hunsberger, Harrysson et al. 2015).
Manufacturing approaches are also stringently governed by current Good Manufacturing Practice (cGMP) (European Commission) standards, US FDA 21CFR and ICH 7-10 to ensure that manufacturing processes are appropriately designed, monitored and controlled under standardized conditions (Skorska, Müller et al. 2017). A cGMP facility is a production facility for the manufacturing of pharmaceutical or cellular products (Giancola, Bonfini et al. 2012). Production must advance through a number of controlled steps supported by quality control monitoring and quality assurance programs — known as a quality management system (QMS) (see Figure 1 below).
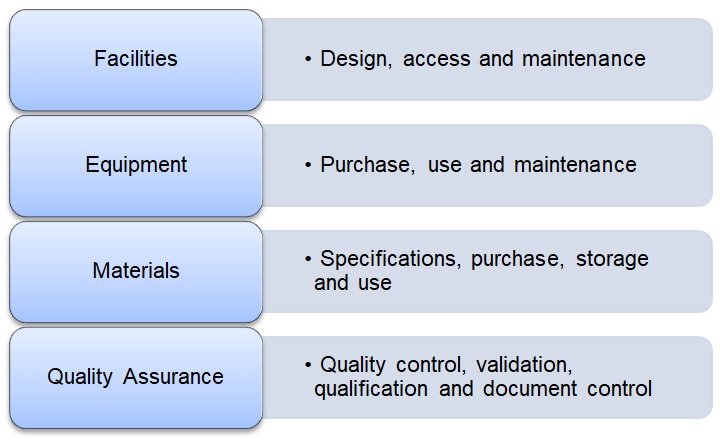
An overview of the Quality System approach that governs the collection, processing, storage and release of cell therapy products (Giancola, Bonfini et al. 2012).
Pertinent steps in the overall cell-based gene therapies manufacturing process include cell isolation, ex vivo cultivation of the target cells followed by genetic modification, expansion and enrichment before re-administration into the patient (see Figure 2 below). As an initial step in this process, packaging of the therapeutic transgene into a delivery vehicle, such as a virus, is required. For this, retroviral or lentiviral vectors are commonly employed, owing to their natural abundance and optimally characterized delivery ability for the transfer of genetic information. Presently, viral vector preference varies although retroviral vectors appear to be more widely used than lentiviral vectors (Brindley, Fuerstenau-Sharp et al. 2016). Significant clinical research efforts are continually attempting to further delineate the benefits of viral vectors and exploit their inherent properties for manufacturing purposes.
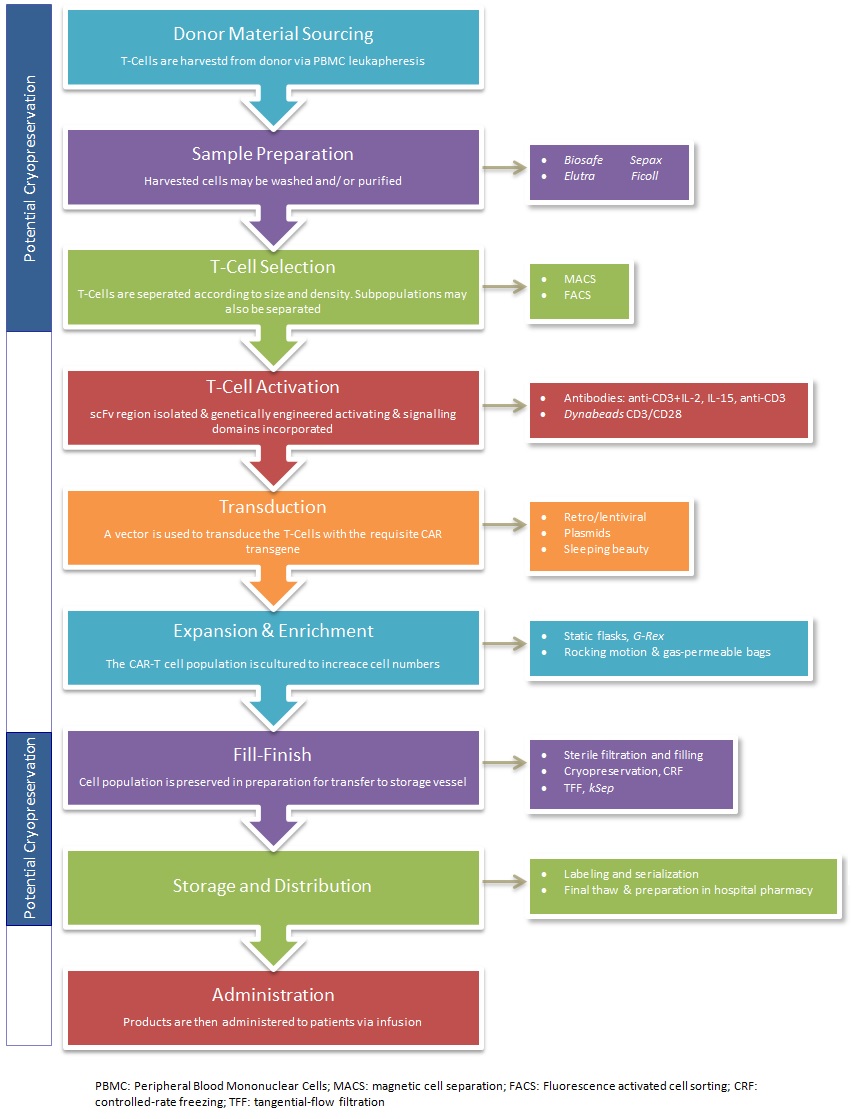
Conventional manufacturing processes for small molecules and biological therapies are well established and these streamlined processes ensure that their production is both sustainable and cost-effective with a long-term reduced cost of goods. Due to the inherent variability of cell therapies, cost-effectiveness remains a key challenge. High startup costs translate into significant barriers-to-entry for novel spinouts and startups, and for established biotech entities, attaining industrial-scale total production yields is challenging. Additional manufacturing challenges relate to the concepts of standardization (with the standardization of cellular input material being especially difficult), and the automation of process steps and workflows.
Conclusion
The global cell therapy community is actively working to address these issues and improve the robustness and quality of manufacturing. Working towards closed systems to reduce contamination risk is aided by commercially available GMP quality culture bags, connectors and accessories, standardization of input cellular material and clearly defined ancillary reagents (Kaiser, Assenmacher et al. 2015). Control of the aforementioned aspects, in addition to simplification of multi-step manufacturing processes (e.g. through automation) will considerably reduce variability, facilitate QC testing and help work towards attainment of cGMP compliant products (Brindley, Wall et al. 2013). Continued efforts to significantly enhance product quality, yield and clinical access, supported by unified regulatory frameworks with expedited approval of promising therapeutic candidates, will help drive cell therapies forwards and cement them as mainstream therapeutic entities.
Acknowledgements
We express sincere thanks to the following organizations that have contributed to the CASMI Translational Stem Cell Consortium (CTSCC; UK) as funding and events partners, without whom the consortium and the benefits it will bring to stem cell translation would be constrained:
GE Healthcare (IL, USA), the Center for Commercialization of Regenerative Medicine (CCRM; CA, USA), Sartorius Stedim Biotech (formerly TAP Biosystems; France), Lonza (Basel, Switzerland), the California Institute for Regenerative Medicine (CA, USA), the Strategies for Engineered Negligible Senescence (SENS) Research Foundation (CA, USA), UK Cell Therapy Catapult (UK), NIH Centre for Regenerative Medicine (MD, USA), the New York Stem Cell Foundation (NY, USA), ThermoFisher Scientific (MA, USA), Eisai (Tokyo, Japan), Medipost (US), Medipost (Korea), Celgene (NJ, USA), Roche (Basel, Switzerland) and Oxford Biomedica (UK).
Financial & Competing Interests Disclosure
DB gratefully acknowledges personal funding from the Oxford Musculoskeletal National Institute for Health Research (NIHR), the Saïd Foundation (London, UK) and the SENS Research Foundation. DB is a stockholder in Translation Ventures Ltd. (Charlbury, UK) and IP Asset Ventures Ltd. (Oxford, UK), companies that among other services provide cell therapy biomanufacturing, regulatory and financial advice to pharmaceutical clients.
DB also is subject to the CFA Institute’s codes, standards, and guidelines, so he must stress that this piece is provided for academic interest only and must not be construed in any way as an investment recommendation. Additionally, at time of publication, DB and the organizations with which he is affiliated may or may not have agreed and/or pending funding commitments from the organizations named herein.
DP gratefully acknowledges support from the CTSCC and the SENS Research Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- Brindley DA, Fuerstenau-Sharp M, Smith JA, Bure K, Pettitt D, Mitrophanous K et al. Emerging Platform Bioprocesses for Viral Vectors and Gene Therapies. Bioprocess Int. 14(Suppl 4), pp. 8-17 (2016)
- Brindley DA, Wall I, Bure K. Automation of cell therapy biomanufacturing. Bioprocess Int. 11(Suppl 3), pp. 18-25 (2013)
- Culme-Seymour EJ, Davie NL, Brindley DA, Edwards-Parton S, Mason C. A decade of cell therapy clinical trials (2000-2010). Regen Med. 7(4), pp. 455-462 (2012)
- DIGIUSTO DL, Kiem HP. Current translational and clinical practices in hematopoietic cell and gene therapy. Cytotherapy. 14(7), pp. 775-790 (2012)
- European Commission. Volume 4 Good Manufacturing Practice. Eudralex Volume 4 Good Manufacturing Practice, 4.
- Giancola R, Bonfini T, Iacone A. Cell therapy: cGMP facilities and manufacturing. Muscles Ligaments Tendons J. 2(3), pp. 243-247 (2012)
- Hunsberger J, Harrysson O, Shirwaiker R, Starly B, Wysk R, Cohen P et al. Manufacturing road map for tissue engineering and regenerative medicine technologies. Stem Cells Trans Med. 4(2), pp. 130-135 (2015)
- Kaiser AD, Assenmacher M, Schröder B, Meyer M, Orentas R, Bethke U et al. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther. 22(2), pp. 72-78 (2015)
- Pettitt D, Arshad Z, Davies B, Smith J, French A, Cole D et al. An assessment of the factors affecting the commercialization of cell-based therapeutics: a systematic review protocol. Syst Rev. 6(1), pp. 120 (2017)
- Skorska A, Müller P, Gaebel R, Grosse J, Lemcke H, Lux CA et al. GMP-conformant on-site manufacturing of a CD133 stem cell product for cardiovascular regeneration. Stem Cell Res Ther. 8(1), pp. 33 (2017)
- Zacharias DG, Nelson TJ, Mueller PS, Hook CC. The science and ethics of induced pluripotency: What will become of embryonic stem cells? Mayo Clin Proc. 86(7), pp. 634-640 (2011)