CRISPR-based gene therapy shows promise for muscular dystrophy

A potential gene-editing approach for treating muscular dystrophy has demonstrated encouraging preclinical results, with hopes of going to clinical trial.
Researchers from the Experimental and Clinical Research Center (ECRC), a joint institution of the Max Delbrück Center and Charité – Universitätsmedizin Berlin (all Berlin, Germany), have developed a gene-editing technique aiming to repair and restore the protein deficiency caused by a mutation associated with muscular dystrophy. The study’s preclinical findings have demonstrated promising results, putting the researchers in a favorable position for potential first-in-human clinical trials.
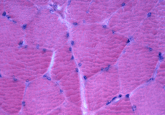
Muscular dystrophy, a group of inherited diseases that cause muscle weakness, affects thousands worldwide. These conditions are often linked to mutations in the DYSF gene, which encodes dysferlin, a protein essential for repairing damaged cell membranes, particularly in muscle fibers. Dysferlin deficiency due to loss-of-function mutations leads to skeletal muscle degeneration and atrophy, including conditions like limb-girdle muscular dystrophy.

A coral-inspired biomimetic material could revolutionize bone repair, closely mimicking natural bone’s structure and biological behavior.
With over 600 muscles in the human body, creating a therapy that targets each muscle is a significant challenge. Simone Spuler, Head of the Myology Lab at ECRC, has dedicated nearly two decades to studying dysferlin. Building on her extensive research, Spuler’s lab, alongside first author Helena Escobar, focused their efforts on addressing a, “more common mutation so that [they] can help as many patients as possible,” according to Escobar.
The team targeted a frameshift loss-of-function mutation in exon 44 of the DYSF gene, which is prevalent in certain patients with limb-girdle muscular dystrophy, and applied CRISPR-Cas9 technology in their study.
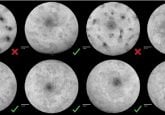
First, they extracted primary muscle stem cells from two patients with limb-girdle muscular dystrophy and used mRNA-mediated delivery of SpCas9 and a mutation-specific single-guide RNA to edit exon 44 in the patient-derived muscle stem cells. Through this gene-editing approach, they re-framed exon 44, correcting the mutation and restoring dysferlin function in cell culture.
Next, the same procedure was replicated in humanized-mice primary muscle stem cells. Through their gene-editing technique, they restored the function of dysferlin and observed muscle regeneration.
Through analysis, the researchers observed reframing efficiencies of more than 60% in exon 44. However, the reframed dysferlin was not identical to the wild-type protein. Despite there being an exchange of four amino acids in the reframed dysferlin, Escobar explained that it “is very similar in function to the wild type, which is the version [seen] in healthy individuals. It localized along damaged cell membranes and muscle was regenerated.”
Furthermore, there were no immune responses detected in the mouse model after assessment, positioning the study for potential human clinical trials.
“We are starting very humbly by targeting one or two muscles. But if this therapy works, it will heal the muscle,” commented Spuler.
The team is now focused on securing funding to begin the first human clinical trial and acknowledges that it may take years before the therapy becomes widely accessible. However, they believe the study’s findings could contribute to the advancement of innovative therapies for dysferlin deficiency-related syndromes and potentially be applied to other forms of muscular dystrophy.